
Research Techniques
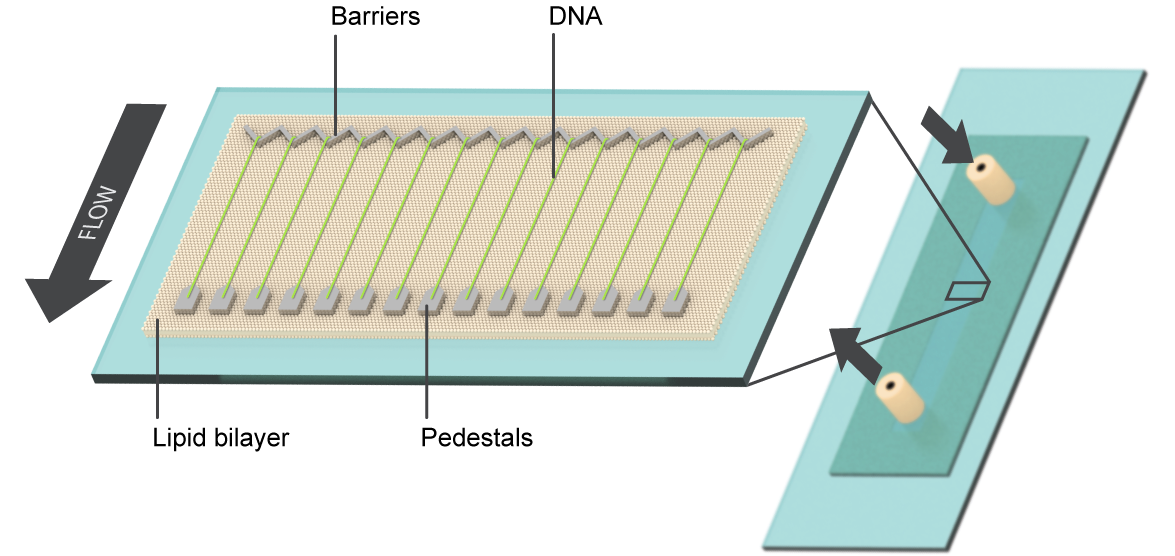

DNA Curtains
This single-molecule optical microscopy technique is used to study fundamental interactions between proteins and nucleic acids, using a non-traditional combination of biochemistry, physics, and nano-scale technology. It allows us to observe individual protein molecules and protein complexes as they interact with their DNA substrates. The goal is to reveal which proteins bind to DNA, where they bind, how they move, and how they influence other components of the system in real-time, on a single-reaction scale.
Our lab developed the DNA curtain technique to overcome limitations on a single-reaction scale in other TIRFM-based biochemistry. The technology uses fluid lipid bilayers to render surfaces inert to biological molecules, and micro- and nano-scale materials engineering to construct parallel arrays containing individual DNA molecules with user-defined positions, orientations, tensions, and topologies. These DNA arrays allow for parallel processing of hundreds to thousands of individual protein-nucleic acid interactions in a single TIRFM experiment and serve as powerful tool for single-molecule research.
Single-particle cryo-electron microscopy
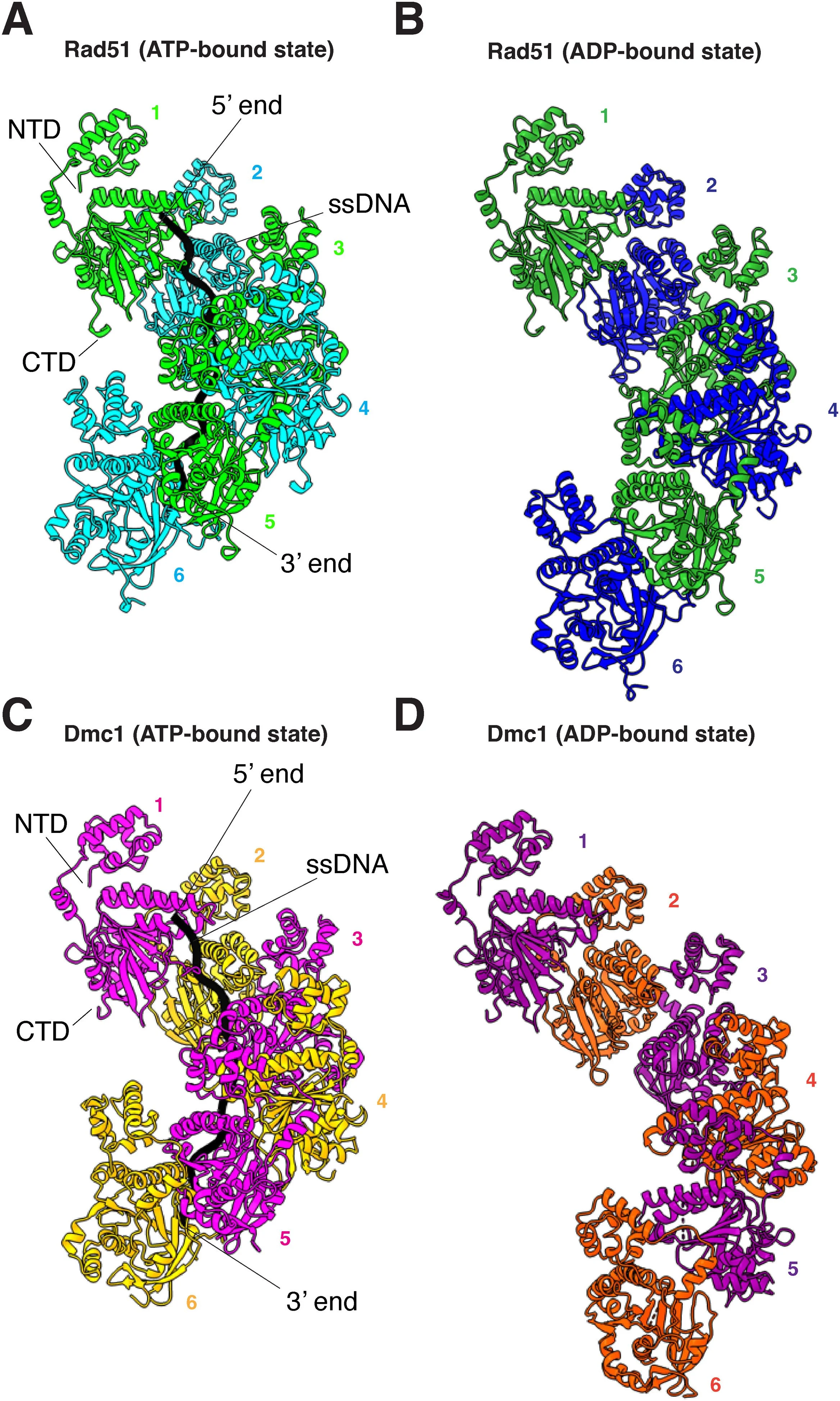
Cryo-electron microscopy (cryo-EM) is a modern structural technique that uses images taken on an electron microscope to reconstruct the 3D structure of proteins and other biological macromolecules. By plunge-freezing samples in liquid ethane, samples freeze without forming ice crystals in a process called vitrification, which preserves molecules in their native state and protects them from being damaged by the electron microscope. Samples are then imaged on an electron microscope, where 2D images are taken of particles in many different orientations that can then be reconstructed into a 3D image.
Our lab uses this technique to understand protein-protein and protein-nucleic acid interactions that occur during the process of homologous recombination. Some of our recent work involving cryo-EM has allowed us to understand the structural transitions that occur within yeast Rad51 & Dmc1 filaments during ATP hydrolysis and how the yeast proteins Rad54 & Hed1 interact with Rad51 filaments.
Recent work involving cryo-EM:
Shin Y, Kim SY, Greene EC. ATP hydrolysis-driven structural transitions within the S. cerevisiae Rad51 and Dmc1 nucleoprotein filaments. J Biol Chem. 2025 Jul 26:110528. doi: 10.1016/j.jbc.2025.110528. Epub ahead of print. PMID: 40721016.
Shin Y, Petassi MT, Jessop AM, Kim SY, Matei R, Morse K, Raina VB, Roy U, Greene EC. Structural basis for Rad54- and Hed1-mediated regulation of Rad51 during the transition from mitotic to meiotic recombination. bioRxiv [Preprint]. 2025 Mar 26:2025.03.26.645561. doi: 10.1101/2025.03.26.645561. PMID: 40196570; PMCID: PMC11974805.
Next-generation sequencing (NGS)
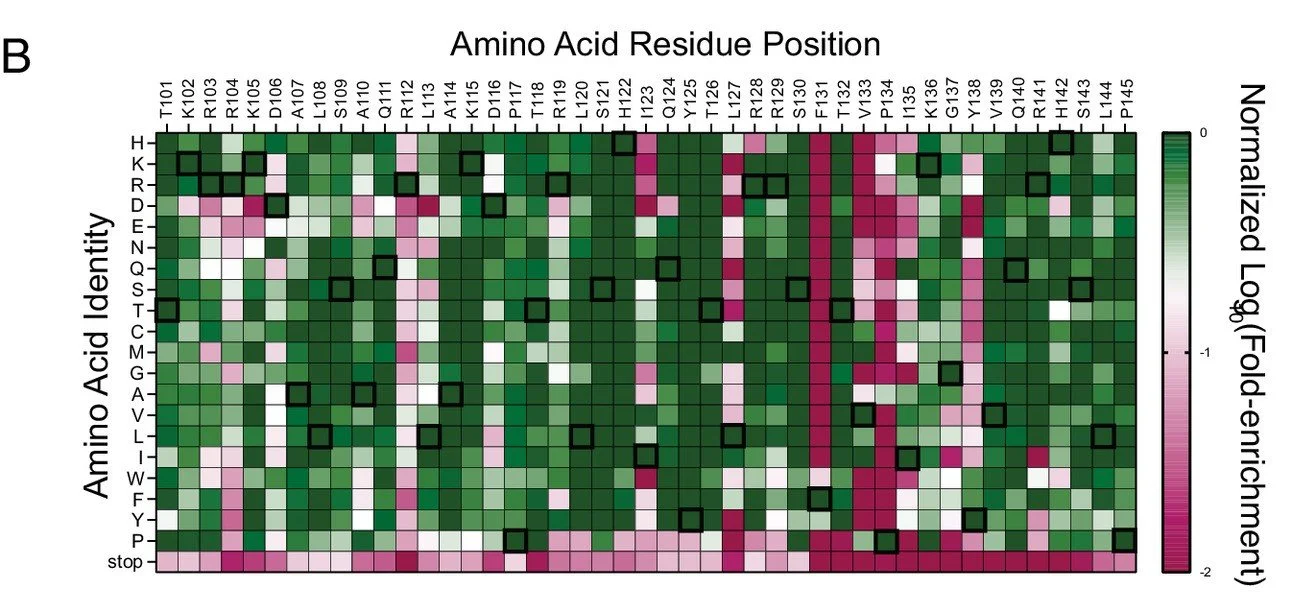
Next-generation sequencing (NGS) is a powerful technology developed for rapid, large-scale genome sequencing, enabling millions of DNA molecules to be read in parallel with exceptional depth and precision. In the Greene lab, we harness this capability to perform high-throughput functional screening of homologous recombination proteins such as Rad51 and Dmc1. By combining deep mutational scanning with selection assays in Saccharomyces cerevisiae, we generate and evaluate thousands of protein variants simultaneously. This approach allows us to quantitatively link sequence variation to protein function, providing detailed insights into DNA repair mechanisms.
Recent work with NGS/Deep Mutational Scanning:
Petassi M, Shin Y, Jessop AM, Morse K, Kim SY, Kuppa S, Matei R, Raina VB, Greene EC. Lineage-specific amino acids define functional attributes of the protomer-protomer interfaces for the Rad51 and Dmc1 recombinases. J Biol Chem. 2026 Jan;302(1):111019. doi: 10.1016/j.jbc.2025.111019. Epub 2025 Dec 8. PMID: 41371341; PMCID: PMC12803942.
Shin Y, Petassi MT, Jessop AM, Kim SY, Matei R, Morse K, Raina VB, Roy U, Greene EC. Structural basis for Rad54- and Hed1-mediated regulation of Rad51 during the transition from mitotic to meiotic recombination. Proc Natl Acad Sci U S A. 2025 Sep 16;122(37):e2510007122. doi: 10.1073/pnas.2510007122. Epub 2025 Sep 11. PMID: 40932772; PMCID: PMC12452912.